

Over the course of nine months, an entire human body is sculpted from a few cells into a baby. The blueprint is the information written into our DNA. But what happens if there is a mistake in these blueprints? Decades worth of research carried out in Malta and abroad have aimed to understand how these errors lead to a disease common in Malta and prevalent worldwide.
Scott Wilcockson talks to Dr Joseph Borg (Faculty of Health Sciences, University of Malta) to find out more.
You are unique. This is not just saying that to make you feel good about yourself. Everyone is unique. Every person is moulded by their upbringing, experiences, and genes. By fusing together their DNA, our parents have made a distinct person with a unique DNA sequence, randomly passing on the best and worst of themselves to the next generation.
The information written into your DNA, or genome, supplies the embryonic ‘you’ with the instructions to build your entire body and then to maintain it throughout your life. While all human beings possess a common set of genes—around 23,000 of them—our DNA is not exactly the same for all of us. We are all riddled with small variations, or mutations, throughout our genomes. Many of these mutations result in biological quirks that play a role in our individuality. For example, mutations in the OCA2 gene are a major determinant of eye colour. Other mutations can have more profound effects on health and well being.
Most diseases have some sort of genetic element, though the exact cause can vary. Some diseases, like diabetes or cancer, are due to many genes malfunctioning and are known as multifactorial. The multiple genetic mutations acting in concert trigger disease progression. Others on the other hand are monogenic as they are triggered by one gene mutation. One such disease is Beta Thalassaemia, which has become the focus of Dr Joseph Borg’s research after he joined Prof. Alex Felice‘s research group that has studied how a genetic quirk could be used to treat this illness.
When blood turns bad
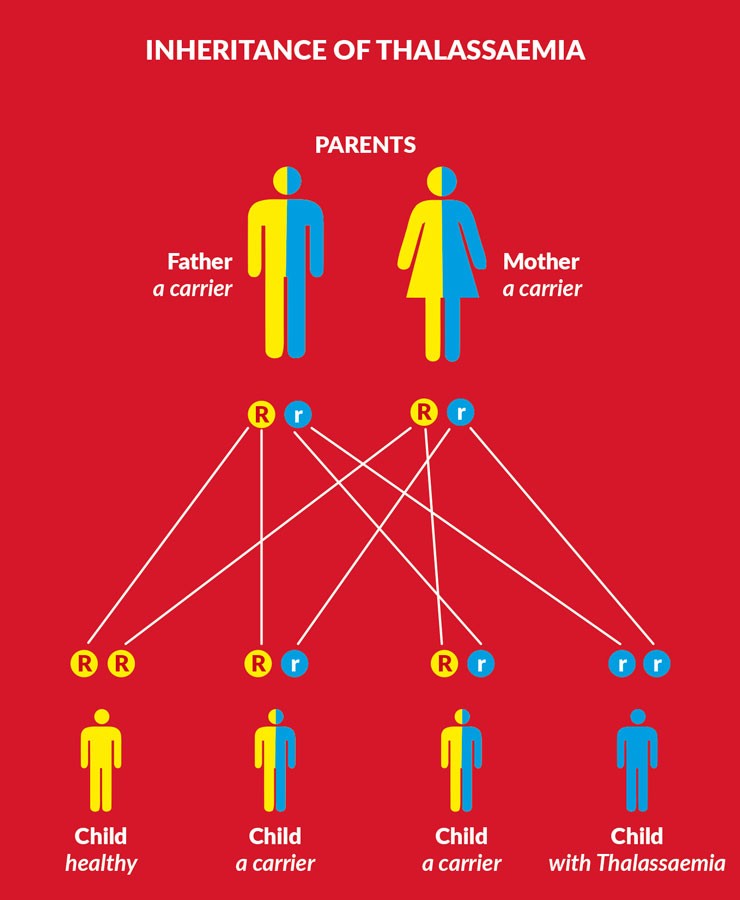
Borg explained that ‘thalassaemia is an inherited blood disorder caused by an inability to produce sufficient amounts of haemoglobin, the [molecule] in red blood cells that binds and transports oxygen to our various tissues and organs.’ The most prominent form in the Mediterranean is Beta Thalassaemia. This is caused by mutations that affect the production of the beta-chain sub-unit of the haemoglobin molecule in blood cells.
There are around 300 known mutations that cause thalassaemia worldwide. In Malta, a single mutation in the beta-globin gene was found to be the main culprit in the late 1980s by Prof. Christian Scerri (Faculty of Medicine and Surgery, University of Malta) and had been designated the very catchy name of β+ IVS-I-6 (T→C). This mutation is common throughout the Western Mediterranean and accounts for two thirds of all Maltese cases.
Unfortunately, treatment options are limited. Borg explains that ‘adult patients have to undergo lifelong blood transfusions every month, or a bone marrow transplant in rare cases.’ Hope is in sight, however. Studies are now focusing on a biological
quirk that could hold the key to treating patients. In the 1990s, a study on the Maltese population carried out by Prof. Alex Felice (Faculty of Medicine and Surgery, University of Malta) and his team identified another common mutation within the gamma-globin gene. This gene contains the blueprints needed to make another haemoglobin protein known as the gamma-chain. In adults the beta-chain is used to make haemoglobin. In babies the gamma-chain is used instead in the developing foetus and up to six months after birth.
“The relative ease with which researchers can sequence all of a person’s genes is paving the way for a much greater understanding of how diseases develop and how to treat them.”
A quirk of nature
When a foetus is developing, oxygen is provided through the mother’s blood supply. Oxygen diffuses through the mother’s placenta in the womb where it is picked up by the baby’s red blood cells. The foetus needs to be able to absorb as much oxygen as possible and therefore uses a different type of haemoglobin (foetal haemoglobin) that has a stronger attraction toward oxygen molecules. Following birth, babies are able to obtain plenty of oxygen on their own and switch from primarily foetal to adult haemoglobin.
Intriguingly, the switch is not always complete. Borg described how a small portion (less than 1%) of all our haemoglobin is foetal even in adulthood. However, some adults can have much higher levels because of certain genetic mutations. This phenomenon is known as Hereditary Persistence of Foetal Haemoglobin (HPFH). Felice first discovered a Maltese person with HPFH, and now whole families have been found.
Earlier, Prof. Maurice Cauchi (now Melbourne, Australia) had discovered the Hb F Malta I variant in the gamma-globin gene. The mutation is found in around 2% of Maltese newborns. Together with other variants they are the most valuable quantitative markers of the foetal to adult globin gene switch unique to Maltese families.
 Individuals who have two mutant genes, one from each parent, can have around 58% of their total haemoglobin to be foetal. One mutant gene leads to between 15–34% foetal haemoglobin. This particular quirk of biology does not cause any ill effects, as foetal haemoglobin functions well and individuals with the mutation usually do not find out until they have it tested.
Individuals who have two mutant genes, one from each parent, can have around 58% of their total haemoglobin to be foetal. One mutant gene leads to between 15–34% foetal haemoglobin. This particular quirk of biology does not cause any ill effects, as foetal haemoglobin functions well and individuals with the mutation usually do not find out until they have it tested.
This biological peculiarity is of great interest to researchers like Felice and Borg. ‘We wanted to use the knowledge of this mechanistic imbalance [in the switch] to understand how best to treat those who suffer from blood disorders like thalassaemia and sickle-cell disease.’ People with these disorders usually have normal foetal haemoglobin but abnormal or insufficient adult haemoglobin. Increasing the level of foetal haemoglobin in adulthood effectively cures these diseases. Borg added how ‘if you can augment higher levels of foetal haemoglobin [in adults] you can render [patients] independent of transfusions. No more treatment. No more medicine. It would give them a better quality of life.’
In 2010, this team published (together with ERASMUS) a seminal work in the world-leading journal Nature Genetics. The work identified a key factor that caused the haemoglobin switch in the first six months of a baby’s life. The study looked at an entire Maltese family, where ten out of 27 members exhibited HPFH with varying degrees of foetal haemoglobin production ranging from 3–20% of the total haemoglobin. Borg and his colleagues sequenced the recruited family’s DNA to identify the genetic mutations causing this imbalance. They found that a single gene called KLF1 was mutated in all family members with HPFH and, to date, this mutation seems Malta-specific.
This gene provides the blueprint to make the KLF1 protein. KLF1 is a transcription factor. It binds to specific parts of the DNA and turns genes on or off. KLF1 switches on genes involved in red blood cell production.
This study revealed how the mutation in the Maltese family blocks the KLF1 protein from binding to DNA and doing its job. KLF1 normally switches on the adult beta-globin gene and turns off the foetal gamma-globin gene. By stopping KLF1 from doing so the Maltese mutation causes a lot more foetal haemoglobin to be produced in adults.
This exciting discovery identified a major regulator of the switch. Borg cautions however, that ‘after our publication, other groups identified similar and also conflicting results because of different KLF1 mutations […]. This [suggested] that KLF1 is not acting alone.’ Another puzzle was that foetal haemoglobin levels varied greatly in family members. They ranged from 17–20% to only 3–5%. The KLF1 mutation alone did not explain the difference, which means it was not capable of driving high foetal haemoglobin levels on its own. Other factors and gene mutations must be at play.
This leads us to the next stage of the research. ‘We have now identified three other Maltese families with the same KLF1 mutation but with normal [foetal haemoglobin] levels, less than 1%.’ Felice, Borg, Ruth Galdies, and their team sequenced all of the DNA (a genome) of over 50 individuals. They are now comparing the genomes of these new families with the earlier study. ‘KLF1 is a common factor [in all families]. We can cancel it out and see what [genes] we are left with. […] We suspect we will be left with the ‘friends’ or ‘foes’ of KLF1 controlling the [foetal haemoglobin] levels.’
The relative ease with which researchers can sequence all of a person’s genes is paving the way for a much greater understanding of how diseases develop and how to treat them.

Mimicking biology
Gene therapy is a possible method to treat diseases like thalassaemia. The idea is to change the gene’s sequence or activity within living people. Borg describes new methods known as gene editing that could be used to correct genetic defects to treat these disease types. ‘This could be used in the form of a vaccine [injected into the patient which] then can home in [and] turn off the [gene].’ Gene therapy is still experimental, due to health and safety concerns, but Borg believes that ‘we are not too far away [from a treatment].’
The advances in gene sequencing technology are rapidly turning science fiction into science fact. Once developed, gene therapy is a giant leap over traditional drugs. ‘What is beautiful is that with today’s technology, you can either choose to fully or partially [turn off a gene],’ Borg explains. ‘This may be very important because the complete absence of a gene can have dire consequences.’ For now, humanity will have to wait till this technology develops.
Luckily, gene sequencing has started to be used alongside conventional medical treatment. Attempts to mimic the effects of HPFH by elevating foetal hemoglobin levels in adults using drugs have been going on for a while. Sickle-cell disease (a disease similar to thalassaemia) is already being treated. The drug Hydroxyurea reactivates the production of foetal haemoglobin in adults and blocks the formation of mutant haemoglobin, which keeps the disease in check.
Hydroxyurea has similarly been tried to treat thalassaemia. The results have been mixed, with some responding better than others. The drug can also have serious side effects and can kill blood cells. It seems that the bone marrow of thalassaemia patients is inflamed and highly sensitive to hydroxyurea. The dosage needs to be carefully regulated to minimise these effects, but also to ensure that individuals are not needlessly exposed to the drug if they do not respond to it. This is where genetics comes back to save the day.
Medicine made just for you…
Personalised medicine is a rapidly growing idea that uses a person’s genetic information to determine the best drug cocktail. By knowing a person’s genetics, an individual’s drug response can be estimated. The right treatment can be matched to the right patient without wasting time and money, while reducing threats to the patient’s health—a dream that needs more research to be realised.
The enormous wealth of information studied by geneticists is migrating from labs into clinics. Thanks to researchers like Felice, Borg, and the rest of the team the ways in which diseases develop are being understood. The aim is to tailor treatments for individual patients. We all share the same 99.9% of DNA. But that 0.1% makes a big difference.
The study’s principal investigator is Prof. Alex Felice with Jeanesse Scerri, Ruth Galdies, and Dr Joseph Borg as close associates.



Comments are closed for this article!